Navigating the development path for FDA-approved companion diagnostics doesn’t have to be complex — it just requires a partner with expertise, experience, and resources who knows the way.
As a Contract Diagnostics Organization (diagnostic-focused CRO), ResearchDx is a unique, integrated business entity that solves logistical complexities associated with multi-partner outsourcing for regulated companion diagnostic development. Unlike traditional development strategies where specialized expertise must be coordinated through multiple vendor organizations, a CDO provides all expertise and services within a single, integrated organization.
By integrating and synchronizing all IVD and biopharma development activities, a single ‘start-to-finish’ partner simplifies and speeds the process to complete your FDA-approved companion diagnostic at the right time.
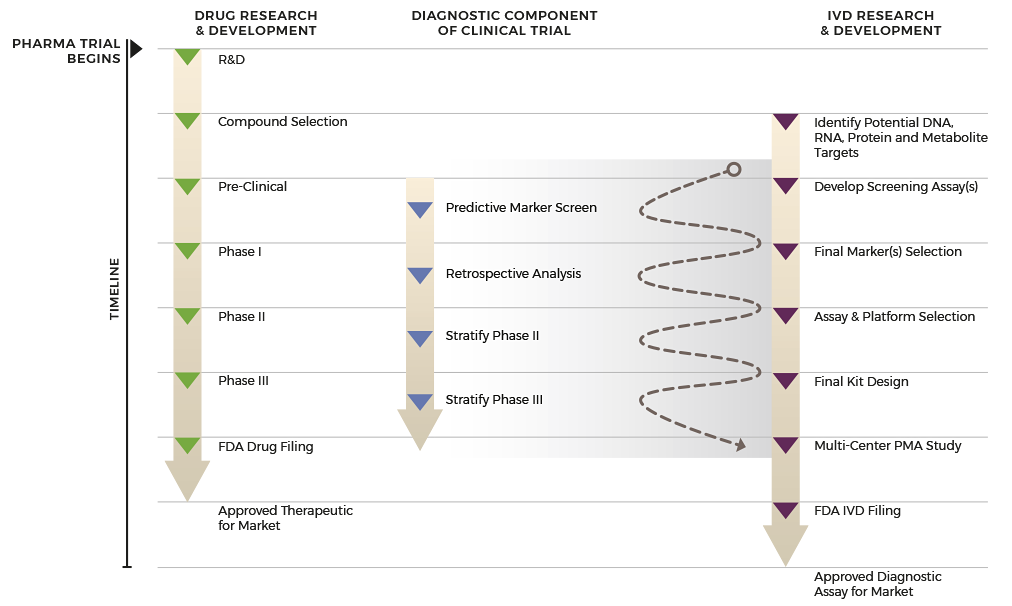
Unlike multi-partner outsourcing, your dedicated project manager at ResearchDx is your point person to guide you through all phases of your project. As an integrated organization, we have built-in flexibility to adapt to your project’s changing needs and priorities with the efficiency of a single-point contact.
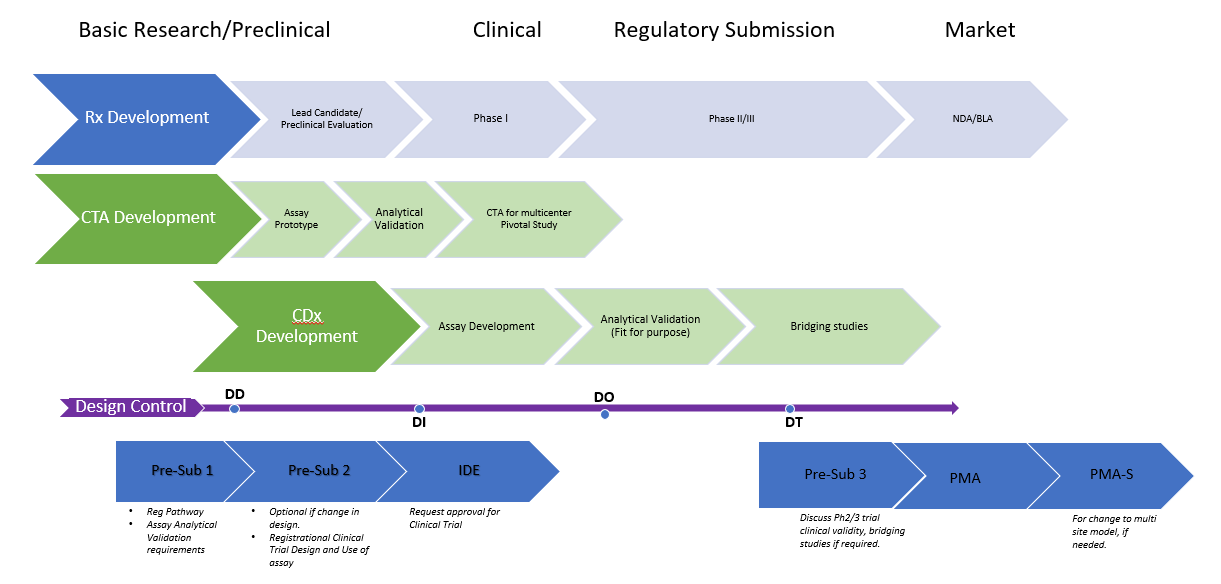
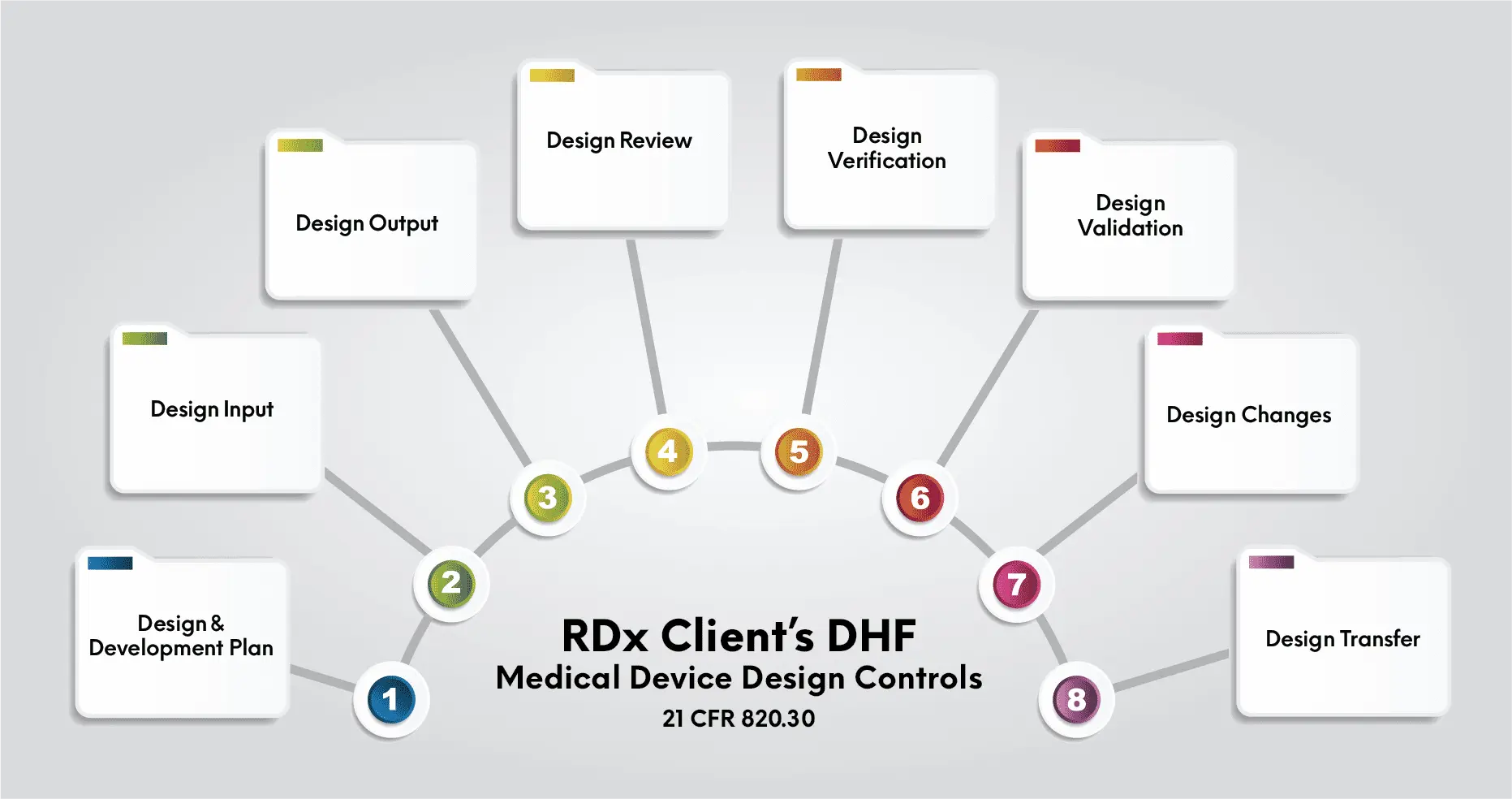
There are two main premarket review pathways for products – premarket notification and PMA.
This is the less stringent of the two and is meant for Class I and some Class II devices similar to existing products on the market.
PMA is a more stringent process that requires the manufacturer to demonstrate the safety and effectiveness of the product.
This pathway is meant for most Class II and all Class III devices as they pose a high risk to patients. It’s also meant for state-of-the-art devices (i.e., devices that aren’t similar to any existing product on the market).